化学者のつぶやき
α,β-不飽和イミンのγ-炭素原子の不斉マイケル付加反応
α,β-不飽和イミンのγ-炭素原子のエナールへのエナンチオ選択的マイケル付加反応が開発された。新規環状β-アミノ酸の有用な合成法となりうる。
遠隔位での不斉マイケル付加反応
不斉アミン触媒を用いたα,β-不飽和カルボニル化合物へのエナンチオ選択的マイケル付加は、強力なキラルC–C結合形成法として研究されてきた。不斉マイケル付加反応![]() の求核剤としてはカルボニル化合物のα位炭素など様々な化学種が知られる。その他の求核剤の一つとして、シリルジエノールエーテルなど、ビニロガスマイケル供与体を用いた遠隔位での不斉誘導反応も近年注目を集めている。不斉アミン触媒を用いたエナンチオ選択的ビニロガス向山–マイケル反応は、MacMillan
の求核剤としてはカルボニル化合物のα位炭素など様々な化学種が知られる。その他の求核剤の一つとして、シリルジエノールエーテルなど、ビニロガスマイケル供与体を用いた遠隔位での不斉誘導反応も近年注目を集めている。不斉アミン触媒を用いたエナンチオ選択的ビニロガス向山–マイケル反応は、MacMillan![]() らによって初めて開発された(図1A)[1]。この反応では、MacMillan触媒存在下、環状ビニロガスマイケル供与体としてα-シリルオキシフランのγ-炭素原子が求核種として働く。しかし、鎖状のビニロガスマイケル供与体を求核剤に用いた報告例はほとんどなかった。2012年にSchneiderらが、林–Jørgensen触媒
らによって初めて開発された(図1A)[1]。この反応では、MacMillan触媒存在下、環状ビニロガスマイケル供与体としてα-シリルオキシフランのγ-炭素原子が求核種として働く。しかし、鎖状のビニロガスマイケル供与体を求核剤に用いた報告例はほとんどなかった。2012年にSchneiderらが、林–Jørgensen触媒![]() 条件下、はじめて鎖状シリルジエノールエーテルを用いたエナンチオ選択的ビニロガス向山–マイケル反応の開発に成功した(図1B)[2]。プロリン触媒による直鎖エナールの二量化反応も知られる(図1C)[3]。これは系中で生じるイミニウムAとエナミンBのビニロガス–マイケル反応経由の付加環化反応である。しかし、収率およびエナンチオ選択性ともに中程度であった。
条件下、はじめて鎖状シリルジエノールエーテルを用いたエナンチオ選択的ビニロガス向山–マイケル反応の開発に成功した(図1B)[2]。プロリン触媒による直鎖エナールの二量化反応も知られる(図1C)[3]。これは系中で生じるイミニウムAとエナミンBのビニロガス–マイケル反応経由の付加環化反応である。しかし、収率およびエナンチオ選択性ともに中程度であった。
今回、南洋工科大学のChi教授らは鎖状のα,β-不飽和イミンをビニロガスマイケル供与体とするエナールへのエナンチオ選択的ビニロガスマイケル付加反応の開発に成功したので紹介する(図1D)。ビニロガスマイケル付加–分子内マンニッヒ反応により環状アミノアルデヒドが得られる。この生成物は容易に環状β-アミノ酸にも誘導できる。
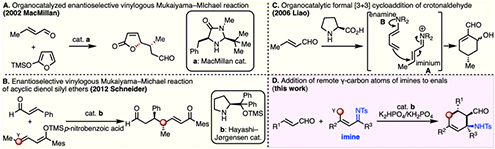
図1. キラルアミンを用いた不斉ビニロガスマイケル反応 (A)環状求核種の例 (B, C)鎖状求核種の例 (D)今回の反応
“
“Access to Cyclic β-Amino Acids by Amine-Catalyzed Enantioselective Addition of the γ-Carbon Atoms of α,β-Unsaturated Imines to Enals”
Luo, G.; Huang, Z.; Zhuo, S.; Mou, C.; Wu, J.; Jin, Z.; Chi, Y.
R. Angew. Chem., Int. Ed. 2019, 58, 17189-17193.
DOI:10.1002/anie.201908896![]()
”
論文著者の紹介

研究者の経歴:
–2002 BSc, Hong Kong Baptist University (HKBU) and Tsinghua University
2002–2007 Ph.D., Department of Chemistry, UW-Madison, USA (Prof. Gellman, S. H.)
2007–2009 Posdoc, Department of Chemistry, UC Berkeley, USA (Prof. Fréchet![]() , J. M. J.)
, J. M. J.)
2009– Assistant Professor, Nanyang Technological University, Singapore
2009– Professor, Nanyang Technological University, Singapore
2012– Distinguished Professor, Guizhou University, China
研究内容:NHCを用いた結合活性化、不斉カルベン触媒反応

研究者:Jian Wu 吴剑![]()
研究者の経歴:詳細は不明
–2010Ph.D, development of fine chemicals of Guizhou University, China
Present:Professor, Guizhou University, China
研究内容:グリーンケミストリー、農薬化学、創薬化学
論文の概要
著者らは、林–Jørgensen触媒、塩基とNaCl存在下、エナール1に対してα,β-不飽和イミン2のγ-炭素がマイケル付加することで、エナンチオ選択的にシクロアミノアルデヒド3を合成できることを見出した(図2A)。推定反応機構は以下の通りである。まず1と林–JØrgensen触媒によりα,β-不飽和イミニウム中間体Iが生成する。一方、塩基が2のγ位C–H結合を脱プロトンし、ジエナミド中間体IIを与える。Iに対し、IIのγ-炭素がマイケル付加することで、中間体IIIとなる。IIIは分子内マンニッヒ反応により、6員環中間体IVとなり、続く加水分解で3が得られる。なお、この環化は平衡であり、熱力学的支配によりジアステレオ選択性が発現する。
エナールはシンナムアルデヒド類が適しており、フェニル基上のパラ位にメトキシ基(3b)、ハロゲン(3c)を有する基質も適用可能である(図2B)。オルト位にメチル基をもつ基質(3d)では、おそらく立体障害により長い反応時間を要し、収率も中程度に留まった。また、シンナムアルデヒド以外にもジエナールを用いても反応は進行した(3e)。アルキル置換エナールでは低収率であった(3f)。α,β-不飽和イミンとしては、β位のフェニル基上にハロゲンを有する基質(3g)やβ位にナフチルを有する基質(3h)でも問題なく反応は進行する。一方、γ位にフェニル基を有する基質では低収率であった(3i)。2の窒素原子上の保護基としては、Ts基だけでなくNs基(3j)やp-Ns基(3k)も適用できた。
著者らはこのp-Ns保護されたアルデヒド3kを用いて、環状β-アミノ酸5の合成に成功した(図2C)。アルデヒド部位の酸化によりカルボン酸4とし、続く保護基の除去により5を合成した。このような環状β-アミノ酸の不斉合成例はなく、本反応の有用性を示唆する結果である。
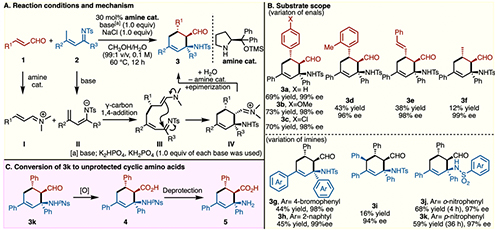
図2. (A)反応条件と推定反応機構、(B)基質適用範囲、(C)生成物の環状アミノ酸への誘導化
以上のように、本論文ではイミンのγ–炭素原子におけるキラルC–C結合形成が達成された。エナンチオ選択的に環状β-アミノ酸を合成できるため、医薬品への応用が期待される。
参考文献
- Brown, S. P.; Goodwin, N. C.; MacMillan, D. W. C. The First Enantioselective Organocatalytic Mukaiyama–Michael Reaction: A Direct Method for the Synthesis of Enantioenriched γ-Butenolide Architecture. J. Am. Chem. Soc. 2003,125, 1192–1194. DOI: 10.1021/ja029095q
- Gupta, V.; Sudhir, S. V.; Mandal, T.; Schneider, C. Organocatalytic, Highly Enantioselective Vinylogous Mukaiyama–Michael Reaction of Acyclic Dienol Silyl Ethers. Angew. Chem., Int. Ed. 2012,51, 12609–12612. DOI: 10.1002/anie.201207058
- Hong, B.; Wu, M.; Tseng, H.; Liao, J. Enantioselective Organocatalytic Formal [3+3]-Cycloaddition of α,β-Unsaturated Aldehydes and Application to the Asymmetric Synthesis of (−)-Isopulegol Hydrate and (−)-Cubebaol. Org. Lett. 2006,8, 2217–2220. DOI: 10.1021/ol060486+
掲載記事について
本記事はWEBに混在する化学情報をまとめ、それを整理、提供する化学ポータルサイト「Chem-Station」の協力のもと、ご提供しております。
お問い合わせ
この製品・ソリューションに関するお問い合わせ、資料請求は、富士フイルム和光純薬(株)までお気軽にお問い合わせください。
- 富士フイルム和光純薬(株) 03-3244-0305