日頃当たり前のように飲んでいる薬。その薬一つひとつが
安心して服用できるよう、「医薬品GMP」という厳しい基準で
製造過程が管理されています。
今回は、このGMPとはどのような内容で、
どのように成立したのかを見ていきましょう。
Reading keywords
安全な薬を作るために!
厳しい基準「医薬品GMP」とは?

医薬品製造の基準、GMPってどんなもの?
薬は私たちの生命に直接関わるもの。そのため調剤や処方の段階はもちろんのこと、医薬品の製造段階でも細心の注意を払う必要があります。日本では、医薬品を製造する際の品質管理の基準はGMP省令と呼ばれる厚生労働省が定める省令で明文化されており、医薬品製造販売の承認を得るためには、医薬品医療機器等法の規定により、多数の資料をそろえてGMP適合性調査を受ける必要があります。
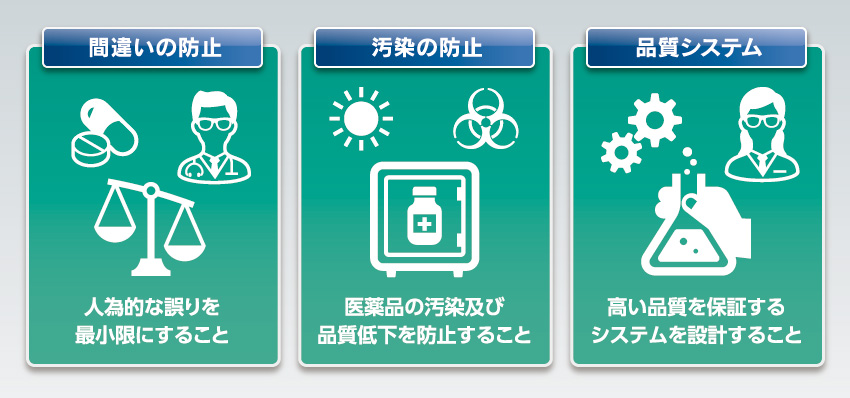
さらに事業を継続させるためには5年ごとの査察を受け、基準を満たしていないと改善指示や指導があるのはもちろんのこと、最悪の場合は業務停止命令を受けることもあるのです。GMPとは、Good Manufacturing Practiceの略。日本語では「医薬品及び医薬部外品の製造管理及び品質管理の基準」と呼ばれます。これは簡単に言うと「患者が安心して使える安全な医薬品を製造するために、医薬品製造所(工場)が守るべき基準」。この省令には、具体的に以下の3つの原則に基づいて記述されています。
- 人為的な誤りを最小限にすること。
- 医薬品の汚染及び品質低下を防止すること。
- 高い品質を保証するシステムを設計すること。
この3原則を実現するために、「ハード(工場の建物や作業室・機械・設備・倉庫の基準)」と「ソフト(組織や基準書・手順書・記録等・管理に関する基準)」の両面からいろいろな決まりを定め、誰が作業を行っても品質に欠陥のない医薬品を作り上げることを実現するのがGMPの意図なのです。
悲劇を繰り返さないために! GMPができたわけ
効き目が高い医薬品ほど人体に強く作用するので、必然的に副作用というリスクをともないます。このため医薬品は時に「もろ刃の剣」となることも。このリスクが問題になった出来事のひとつに「サリドマイド事件」があります。
1957年に西ドイツで開発されたサリドマイドという薬は、もともと副作用が少ない睡眠薬・鎮痛剤として日本を含む世界の国々で使用されていました。ところが、1961年に妊婦が服用すると胎児の手足などに先天異常が生じたり、胎児の死亡を引き起こしたりすることが明らかとなったのです。世界で広く使われていたサリドマイドは多くの被害者を出し、日本でもおよそ1,000人のサリドマイド被害者が出る結果となりました。
この事案は、世界中で医薬品の安全性や有効性を確保することの重要性があらためて見直されるきっかけとなりました。アメリカの厚生労働省にあたるFDA(Food and Drug Administration : 米国食品医薬品局)は、1962年に高品質な医薬品を製造するために必要な設備構造、生産管理、品質管理等に関する基準を「薬品の製造規範に関する事項」として制定。これがGMPの始まりとなります。
その後、WHO(世界保健機関)がアメリカの基準をベースにWHO-GMPを作成し、1969年に国連総会で採択され、実施が加盟国に勧告されました。日本ではこの勧告を受け、1976年より「医薬品の製造および品質管理に関する基準」に基づく行政指導を開始し、その後、1980年に厚生省令として「医薬品GMP」が施行されました。1994年の薬事法改正ではこの「医薬品GMP」が医薬品製造の許可要件となり、さらに 2005年には製造販売の承認要件となりました。
なお、アメリカではcGMP、欧州ではEU GMP、韓国ではK-GMPなど、各国がそれぞれ自国の基準としてGMPを導入しています。これらの基準には細かい違いがあり、特にグローバル企業は国ごとに別の対応をする必要があるので、欧州各国の薬事行政当局を中心に設立されたPIC/Sという団体が、標準化のための話し合いを行っています。
変化する時代に対応するGMP
GMPは一度決まったものを変わらず順守していけばいい、というものではなく、時代や環境・技術の変化にあわせて何度も改正が行われています。例えば、1970年代のアメリカで、輸液の滅菌が不完全だったことにより死亡事故が発生したのをきっかけとして、1976年の改正時に「バリデーション(Validation)」という概念が、アメリカのGMPであるcGMPに取り入れられました。
バリデーションとは、医薬品・医療機器を製造する工程や方法が正しいかどうか、科学的根拠や妥当性があるかどうかを調査・検証するための一連の業務のことです。この概念は日本のGMPにも1995年に導入され、2013年にはバリデーションの基準について一部改定が行われています。このように、日本のGMP省令も時代の要求にあわせて変化してきているのです。
また、2018年には日本と欧州連合(EU)の間でGMPの相互承認協定が結ばれるという進展もあり、医薬品をめぐる周辺業界からは注目が集まっています。医薬品の製造会社は、今後もアップデートされるGMPを順守し、安全で責任のある薬剤製造を行っていくことが期待されています。
「医薬品GMP」への富士フイルムの取り組み
-
- 医薬品GMPに対応した医薬品中間体・原薬の受託製造
- 富士フイルムがこれまでに蓄積したファインケミカル合成技術を生かして構造が複雑化している最近の医薬品の合成に対応すべく研究を重ね、実績をあげています。この経験と実績を活かし、どのような有機合成でもご相談に応じます。
- 医薬品GMP設備

記事公開:2020年3月
情報は公開時点のものです