病気に悩む方々にとって、新しい治療薬の登場はまさに希望です。しかし、日本では
海外で承認を受けた革新的な新薬が使えるようになるまでに、長い時間がかかる
ことが指摘されてきました。さらに最近では、海外で利用されているのに日本では
未承認のため利用することができないことも。薬はあるのに使えない──。
今回はそんな「ドラッグ・ラグ」「ドラッグ・ロス」について解説します。
Reading keywords
海外で使える新薬が国内では使えない!?
深刻化する「ドラッグ・ラグ/ドラッグ・ロス」とは。

新薬の利用が遅れる
「ドラッグ・ラグ」
新薬が開発されて海外では承認されているにもかかわらず、日本では使えない。そこには、「ドラッグ・ラグ」と「ドラッグ・ロス」という2つの問題があります。順番に見ていきましょう。
「ドラッグ・ラグ」とは、海外で承認された新薬が日本で使えるようになるまでに、長い時間がかかってしまう問題です。例えば2010年の時点では、ある医薬品が世界で初めて発売されてから各国で発売されるまでの平均期間はアメリカ0.9年、イギリス1.2年、ドイツ1.3年とおおむね1年前後でしたが、日本では実に4.7年も要していました。
ドラッグ・ラグは、大きく2つのラグによって生じます。1つは「開発ラグ」。これは、海外での治験開始と日本での治験開始の時間差です。治験とは、新しい医薬品や治療法の安全性や有効性を、ヒトを対象に評価する臨床試験のこと。医薬品が市場に出る前に実際の患者に使用して効果や副作用を確かめる重要なプロセスのひとつです。もう1つは「審査ラグ」。開発企業が新薬の製造販売承認申請をした後、各国の規制当局による審査が完了するまでの期間の差です。この2つのラグの合計がドラッグ・ラグとなります。
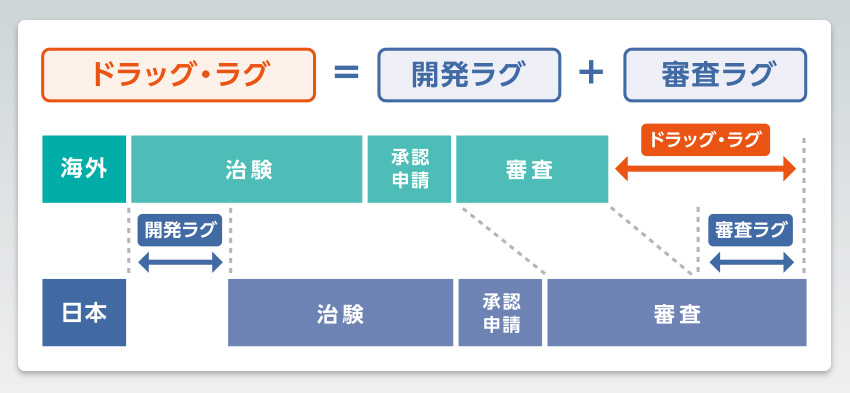
日本においては国、製薬企業、そして国から医薬品の審査などを委託されている独立行政法人 医薬品医療機器総合機構(PMDA/日本における薬事規制当局)では、承認審査プロセスの迅速化を図るため、次のような取り組みを進めています。
- ・国際共同治験の促進
- 治験を複数の国や地域で同時に実施することによって、医薬品の開発と承認申請を海外と同時に行い、日本の治験の開始時期を早めることが目的です。
- ・治験・臨床研究ネットワーク体制の推進
- 海外と比べて国内の病院は規模が小さく、治験の実施が分散化して非効率的でした。そこで専門性と効率化に必要な機能を備えた中核となるコアセンターを設置し、複数の病院をネットワーク化することで、治験にかかる時間の短縮を図っています。
- ・PMDA審査員の増員
- PMDAでは承認審査を行う人員を増やし、それぞれが経験を積むことで審査の迅速化を図っています。また、医薬品の開発段階から製薬企業と治験方法についての相談を充実させています。
このような取り組みの結果、開発ラグと審査ラグを合計したドラッグ・ラグは2018年度の0.9年から2022年度では0.4年にまで短縮されています。しかし、その一方で深刻な課題が新たに浮上してきました。それがもう1つの問題である「ドラッグ・ロス」です。
新薬を利用できない
「ドラッグ・ロス」
「ドラッグ・ロス」とは、欧米などの海外ではすでに使われている治療薬が、日本では開発着手されていない、つまり製薬企業に開発する予定がなく、利用できない状態のこと。2023年3月時点で、欧米では承認されているものの国内での開発未着手の医薬品は86品目あり、未承認薬143品目の60.1%にあたります。さらに、86品目のうち39品目は日本にその病気に対する既存薬がないことを、中央社会保険医療協議会の薬価専門部会が報告しています。特定の病気に限らず、さまざまな領域で「欧米の薬が日本で使えない」状態が発生しているのです。
ドラッグ・ロスが深刻化している背景のひとつが、日本の薬価制度だといわれています。薬価制度とは、国の医療保険で使える医薬品の価格を決める仕組みのこと。日本では2年に1回、市場の実勢価格に合わせて薬価が改定され、多くの場合、改定されるたびに引き下げられてきました。しかし薬価引き下げは医療費の抑制や患者負担の軽減につながる一方、製薬企業にとっては日本市場の魅力を低下させる要因にもなります。
特に、希少疾病や小児、難病などを対象とする医薬品は、市場規模が小さく、日本での売り上げ見込みが低いとされ、国内での開発が未着手の割合が高くなっています。また、日本での薬事承認には、日本人を対象とした臨床試験での有効性と安全性の検証が必要であり、これが追加の負担となっていると指摘されています。
最近では、創薬ベンチャー企業が対象患者数の少ない病気に対する治療薬の開発に挑戦し、大手製薬企業がその成果を買い上げるケースも増えています。そういった場合、資金が限られるベンチャー企業は市場規模の大きな欧米市場を優先し、薬価改定によって収益に不確実性のある日本市場を後回しにしたり、避けたりする傾向も見られます。
ドラッグ・ロスによって最新の革新的な治療薬が日本で使えなくなることは、患者に深刻な影響を及ぼします。最大の影響は、治療の機会を逸してしまう可能性です。もし未承認薬を使えたとしても、保険が適用されず全額自己負担となるため莫大な医療費負担がのしかかります。また、適正に使用した医薬品による健康被害に対しては「医薬品副作用被害救済制度」によって医療費や年金などの給付を受けられますが、未承認薬による健康被害には適用されません。
日本で使えない薬を
減らすための取り組み
ドラッグ・ロス対策についても、すでにさまざまな手が打たれています。
2009年に未承認薬の開発支援を目的として一般社団法人 未承認薬等開発支援センター(現 新薬・未承認薬等研究開発支援センター)が設立され、未承認薬などの研究開発や承認取得などに対する支援などを行っています。
2016年には、「患者申出療養」制度を創設。これは患者からの申し出を受けて未承認薬などの先進的な医療を、できるだけ身近な医療機関で「保険外併用療養費」として受けられるようにするための制度です。未承認薬や入院料などが保険給付対象になるため、自己負担額も軽減されます。
また2023年には、「可能な限り日本人における薬物動態等に関する情報を収集することが望ましい」ことも明確化した上で、国際共同治験の際に日本人で安全性を確かめる事前試験を原則不要とする方針を厚生労働省が示しました。日本独自のルールが見直され、国際共同治験に関する実施条件が大幅に緩和されました。
さらに日本市場への参入インセンティブを高めるため、2014年度の薬価制度改革で世界に先駆けて日本で薬事承認を取得した革新的な医薬品を経済的に評価する「先駆導入加算」が導入されました。2024年度からは「迅速導入加算」が新設され、日本での治験開始が海外と同等もしくは先行する新薬については、薬価に一定の加算が行われます。
しかし、ドラッグ・ラグやドラッグ・ロスを解消するためには、さらなる戦略的な取り組みが不可欠です。そのひとつが、国際共同治験の推進や治験環境の整備です。日本の治験コストが国際的に高く、また日本人症例の組み入れのタイミングが他国の動きと合わず、国際共同治験から日本を外す動きも見られます。データの一元管理や治験の電子化、臨床試験への参加の改善などにより、日本での治験のパフォーマンスを向上させ、国際的なポジションを向上させる取り組みが求められます。
医薬品は私たちの健康維持と医療の質の向上に不可欠な存在です。治療できる医薬品が存在するのであれば、早く確実に患者の手に届くことが求められます。革新的な治療薬を待ち望む多くの命のために、一刻も早いドラッグ・ラグやドラッグ・ロスの解消が望まれます。
「製薬」に関する富士フイルムのサービス
-
- 創薬支援・再生医療
- 新薬の研究開発から生産、再生医療の領域で、細胞・培地・試薬を合わせたソリューションを提供し、アンメットメディカルニーズへの対応に貢献します。
- 製品/ソリューション

-
- 製薬企業向けBPOサービス
- 臨床開発から市販後調査まで、製薬企業向けの業務効率化につながるBPOサービスを提供します。
- サービス紹介

記事公開:2024年4月
情報は公開時点のものです